Studienaktivitäten der GSG-MPN
- GSG-MPN-Register
- FAMy-Studie (rekrutiert)
- FRACTION-Studie (rekrutiert)
- Ruxo-BEAT-Studie (gestoppt)
- POMINC- (MPNSG-0212-) Studie (geschlossen)
- RuxoAllo-Studie (geschlossen)
Im Rahmen der GSG-MPN werden Investigator-initiierte Studien (sog. IITs) durchgeführt, um Patienten innovative Therapien zur Verfügung stellen zu können. Darüber hinaus erlauben IITs die Bearbeitung wissenschaftlicher Fragestellungen und können damit auch zur Klärung beitragen, welche Patientengruppen von der Studientherapie besonders profitieren. Diese Studien stellen daher sowohl für MPN-Patienten als auch für die Inhalte und Ziele der GSG-MPN eine sehr wertvolle Ressource dar. Zum aktuellen Zeitpunkt stehen drei unterschiedliche IITs für ET-, PV- bzw. MF-Patienten mit jeweils bestimmten Erkrankungskonstellationen zur Verfügung, die im nachfolgenden näher ausgeführt werden. Der Abb. 1 ist eine graphische Zusammenstellung der derzeitigen Studienaktivitäten zu entnehmen. Eine Teilnahme an einer IIT der GSG-MPN nur möglich ist, wenn ein Patient zuvor der Teilnahme am Deutschen MPN-Register („Deutsches MPN-Register und Biomaterialbank für BCR-ABL1-negative myeloische Neoplasien“) zugestimmt hat. Teilnehmende Zentren und Ansprechpartner der IITs sind unter „Zentren der GSG-MPN“ gelistet.
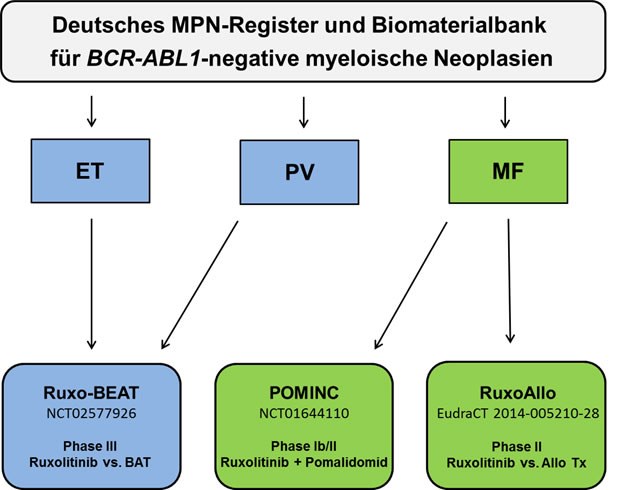
Abb. 1: Übersicht über die derzeitigen Studienaktivitäten der GSG-MPN. Als Dach fungiert das „Deutsche MPN-Register und Biomaterialbank für BCR-ABL1-negative myeloische Neoplasien“, dem Patienten zustimmen müssen, um in eine der drei Therapiestudien (Ruxo-BEAT, POMINC oder RuxoAllo) eingeschlossen werden zu können.
FAMy-Studie (Phase I)
Leitung
Prof. Dr. med. Haifa Kathrin Al-AliUniversitätsmedizin Halle (Saale)
Krukenberg Krebs Zentrum
Ernst Grube-St. 40
06120 Halle (Saale)
Deutschland
Telefon: 0345-5574909
Fax: 0345-5577720
E-Mail: haifa.al-ali@uk-halle.de
Ansprechpartner
Koordinierungszentrum für Klinische Studien (KKS Halle)Medizinische Fakultät der Martin-Luther-Universität Halle-Wittenberg
06097 Halle (Saale)
Deutschland
Telefon: 0345/5574907
Fax: 0345/5575210
E-Mail: famy@kks-halle.de
Die FAMy-Studie ist eine multizentrische, offene Phase 1/2 Studie zur Beurteilung der Wirksamkeit und Sicherheit von Fedratinib in Kombination mit CC-486 bei Patienten mit Myelofibrose in akzelerierter Phase. Die Primären Endpunkte der Phase I sind die Bestimmung der Sicherheit und Verträglichkeit von Fedratinib in Kombination mit CC-486, sowie die Bestimmung der maximal verträglichen Dosis (MTD) von Fedratinib in Kombination mit CC-486. In der Phase II stellt die Ansprechrate (best response) in Woche 24 der Kombinationsbehandlung den primären Endpunkt dar. Weiterhin werden im Rahmen der Begleitforschung umfassende genomische und molekulare Untersuchungen durchgeführt. Die geplante Fallzahl in Phase I und II umfasst 44 Patienten.
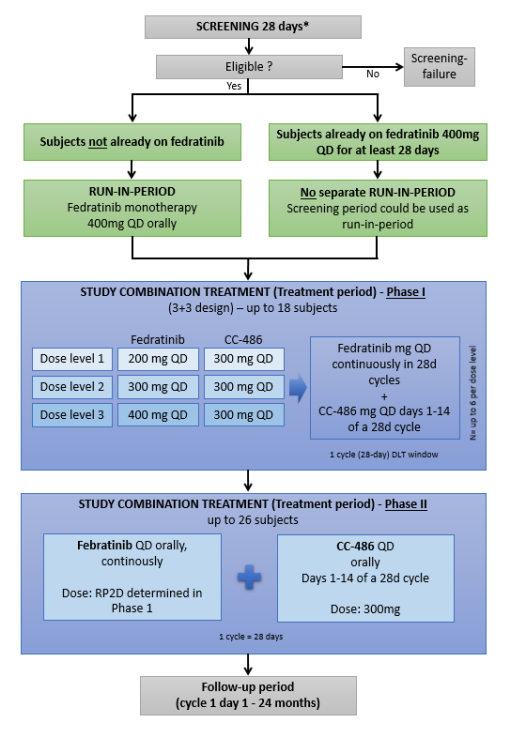
Einschlusskriterien:
1. Alter zum Zeitpunkt der Unterzeichnung der Einverständniserklärung (ICF) mindestens 18 Jahre2. Unterzeichnete Einverständniserklärung liegt vor
3. Diagnose einer MPN-AP (definiert durch eine Blastenzahl von 10%-19% im peripheren Blut oder im Knochenmark) zum Zeitpunkt der Diagnose oder zu einem beliebigen Zeitpunkt im Verlauf einer bekannten primären Myelofibrose (PMF), Myelofibrose (MF) nach Polycythemia vera (PV) oder MF nach essentieller Thrombozythämie (ET)
4. ECOG Performance Score von 0, 1 oder 2
5. Vor Tag 1 der Studientherapie müssen alle behandlungsbedingten Toxizitäten einer vorherigen Therapie auf Grad 1 oder den Ausgangswert der letzten Therapie abgeklungen sein.
Ausschlusskriterien:
1. Patienten mit Thiaminmangel, definiert als Thiaminspiegel unterhalb des Normalbereichs nach lokalem Standard und nicht nachweislich korrigiert vor Beginn der Studienbehandlung.2. Bekannte oder vermutete Überempfindlichkeit gegenüber Azacitidin, Mannitol oder seinen Bestandteilen
3. Patienten, die bekanntermaßen eine Tagesdosis von 400 mg Fedratinib nicht vertragen
4. Patienten mit folgenden Laborwerten innerhalb von 14 Tagen vor der ersten Dosis:
a. Thrombozytenzahl < 1,0 x 109/L L
b. Leukozyten (WBC) > 100 x 109/L L
c. Myeloblasten ≥ 20 % im peripheren Blut
d. Serumkreatinin > 2,5 x obere Grenze der Norm (ULN)
e. Amylase oder Lipase im Serum > 1,5 x ULN
f. Aspartat-Aminotransferase (AST) oder Alanin- Aminotransferase (ALT) > 2,5 x ULN
g. Gesamtbilirubin > 1,5 x ULN, Probanden mit einem Gesamtbilirubin zwischen 1,5 - 3,0 x ULN sind geeignet, wenn der Anteil an direktem Bilirubin < 25 % des Gesamtbilirubins beträgt
5. Patienten mit einer stattgehabten Enzephalopathie, einschließlich Wernicke Encephalopathie (WE)
6. Patienten mit Anzeichen oder Symptomen einer Enzephalopathie, einschließlich WE (z. B. schwere Ataxie, Augenlähmung oder zerebelläre Anzeichen)
7. Patienten mit gleichzeitiger Behandlung oder Verwendung von Arzneimitteln, pflanzlichen Wirkstoffen oder Nahrungsmitteln, die bekanntermaßen starke Induktoren von Cytochrom P450 3A4 (CYP3A4) sind, oder duale CYP2C19- und CYP3A4-Inhibitoren
8. Patienten unter fortlaufender aktiver Therapie gegen ihre MPN-AP (u.a. Chemotherapie, hypomethylierenden Substanzen (HMA), immunmodulatorischer Arzneimitteltherapie (z. B. Thalidomid, Interferon-alpha), Anagrelid, immunsuppressiver Therapie, systemische Kortikosteroide > 10 mg/Tag Prednison oder Äquivalent).
9. Diagnose einer aktiven Lebererkrankung (z. B. chronische alkoholische Lebererkrankung, Autoimmunhepatitis, sklerosierende Cholangitis, primär biliäre Zirrhose, Hämochromatose, nichtalkoholische Steatohepatitis)
10. Patienten mit einer in den letzten 3 Jahren vor Studieneinschluss therapiepflichtigen malignen Erkrankung, die nicht die in der Studie untersuchte Erkrankung ist.
11. Signifikante aktive Herzerkrankung innerhalb der letzten 6 Monate, einschließlich: a. Herzinsuffizienz New York Heart Association (NYHA) Klasse IV b. Instabile Angina pectoris oder Angina pectoris, die eine chirurgische oder medizinische Intervention erfordert; und/oder c. Myokardinfarkt
12. Patienten mit Magenerkrankungen oder anderen Störungen, die zu einer Malabsorption oraler Medikamente führt
13. Patienten, die nicht in der Lage sind, die Kapsel zu schlucken
14. Schwangerschaft oder Stillzeit
15. behördlich und gerichtlich untergebrachte Personen
FRACTION-Studie
Ansprechpartner
Frankfurter Institut für Klinische Krebsforschung IKF GmbH
am Krankenhaus Nordwest
Steinbacher Hohl 2-26
60488 Frankfurt am Main
Germany
Prof. Dr. med. Florian Heidel
E-Mail: heidel.florian@mh-hannover.de
Telefon: 0511 532 ext 3020
Fax: 069 76 01 – 36 55
Luisa Wohn
E-Mail: wohn.luisa@ikf-khnw.de
Telefon: 0 69 5899 787 - 76
Die FRACTION-Studie wird in einer offenen, einarmigen Phase-II-Studie die klinische Wirksamkeit der Kombinationstherapie aus Fedratinib und Nivolumab bei Patienten mit primärer und sekundärer Myelofibrose auf der Grundlage der Konsenskriterien der
Einschlusskriterien:
Die Patienten müssen ALLE der folgenden Einschlusskriterien erfüllen, um für die Studie in Frage zu kommen:a. Unterzeichnete Einverständniserklärung liegt vor
b. Patient*innen ≥18 Jahre alt
c. Patienten, bei denen eine Myelofibrose (MF) gemäß den WHO-Kriterien von 2008 oder 2016 diagnostiziert wurde, einschließlich primärer (präfibrotischer oder offener) und sekundärer Myelofibrose.
d. Patienten mit einer Indikation für eine Therapie (entweder symptomatische Patienten mit Splenomegalie >11cm Durchmesser und/oder Symptomen, die ihre täglichen Aktivitäten einschränken, oder Patienten mit DIPSS int-2, oder hohem Risiko oder MIPSS70 int oder hoch)
e. Patienten, die auf eine JAK-Inhibitor-Therapie nicht oder nur suboptimal ansprechen (in Bezug auf persistierende Symptome, Splenomegalie, Zytopenie oder Hyperproliferation), definiert entweder durch
- Persistierende Splenomegalie >11cm Gesamtdurchmesser
- ODER Anhaltende Leukoerythroblastose
- ODER Anämie <6,2 mmol/l (<10g/dl)
- ODER Erhöhte WBC (>11 Gpt/l)
- ODER Persistierende allgemeine oder konstitutionelle Symptome (Persistenz ist definiert als eine Verringerung um weniger als 50 % gegenüber dem Ausgangswert bei Verwendung des MPN10-TSS-Scores)
- ODER Versagen [sekundäre Resistenz] der JAK-Inhibitor-Behandlung gemäß den IWG-MRT-Kriterien.
f. ECOG-Leistungsstatus <3 beim Screening und ausreichende Organfunktion
g. Zuverlässige Empfängnisverhütung sollte während der gesamten Studie und für 1 Monat nach Absetzen von Fedratinib oder 5 Monate nach Absetzen von Nivolumab** beibehalten werden
h. Der Proband muss bereit sein, eine Transfusion von Blutprodukten zu erhalten
i. Thiaminspiegel nicht unterhalb des Normalbereich (vorherige Substitution möglich)
j. Normaler Ernährungszustand, im Ermessen des behandelnden Arztes
k. Frauen im gebärfähigen Alter (FCBP) müssen sich einem wiederholten Schwangerschaftstest (Serum oder Urin) unterziehen, und die Ergebnisse müssen negativ sein.
l. Sexuell aktive Frauen im gebärfähigen Alter müssen sich verpflichten, angemessene Verhütungsmethoden anzuwenden (d. h. eine Versagensrate von < 1 % pro Jahr), es sei denn, sie praktizieren völlige Abstinenz vom heterosexuellen Verkehr.
m. Männer (einschließlich derjenigen, die eine Vasektomie hinter sich haben) müssen bei sexuellen Aktivitäten mit FCBP Barriere-Verhütungsmittel (Kondome) verwenden. Männer müssen sich verpflichten, kein Sperma oder Samen zu spenden
*Es gibt keine Daten, die auf eine spezielle Geschlechtsverteilung hinweisen, und das Risiko, an Myelofibrose (MF) zu erkranken, hängt nicht vom Geschlecht des Patienten ab. Daher werden die Patienten geschlechtsunabhängig in die Studie aufgenommen.
Ausschlusskriterien:
Das Vorhandensein von JEDEM der folgenden Kriterien schließt einen Patienten von der Teilnahme an der Studie aus:a. Geplante hämatopoetische Stammzelltransplantation innerhalb von 3 Monaten und geeigneter Spender verfügbar
b. >10% Blasten im Knochenmarkausstrich (Zytologie) oder >2x im Blutausstrich während der Screening-Phase oder >20% Blasten zu irgendeinem Zeitpunkt im Knochenmark oder im peripheren Blutausstrich
c. Kreatinin >2xN und Kreatinin-Clearance <45ml/min; ALAT, ASAT & Bilirubin >3xN (wenn MF-Auswirkung auf die Leber >5xN)
d. Diagnose von PV, ET (gemäß WHO 2016) oder positiver molekularer Test auf BCR-ABL
e. Patienten, die laufend Medikamente gegen Myelofibrose einnehmen, einschließlich systemischer Kortikosteroide (eine detaillierte Liste der zulässigen Medikamente ist in Abschnitt 8.2.6.4 und Anhang IV enthalten). Einnahme von systemischen Steroiden innerhalb von 14 Tagen vor der ersten Verabreichung des Prüfpräparats oder während der Studie ist verboten.
f. Unkontrollierte Infektionen
g. Aktuelle Teilnahme an einer anderen interventionellen klinischen Studie innerhalb von 30 Tagen vor der ersten Verabreichung des Prüfpräparats oder zu irgendeinem Zeitpunkt während der Studie
h. Keine Einwilligung zur Erfassung, Speicherung und Verarbeitung der individuellen Krankheitsmerkmale und -verläufe sowie Information des Hausarztes über die Studienteilnahme
i. Keine Einwilligung zum Biobanking von biologischen Proben des Patienten
j. Vorherige Therapie mit Checkpoint-Inhibitoren
k. Impfung innerhalb von 4 Wochen vor Beginn der Behandlung
l. Autoimmunerkrankungen in der Vorgeschichte oder unkontrollierte Autoimmunerkrankungen wie Autoimmunhepatitis, -pneumonitis, -thyreoiditis, chronisch entzündliche Darmerkrankungen, Multiple Sklerose oder rheumatologische Erkrankungen (einschließlich, aber nicht beschränkt auf systemischen Lupus und Vaskulitis)
m. Bösartige Erkrankungen in der Vorgeschichte mit Ausnahme von i) angemessen behandeltem lokalem Basalzell- oder Plattenepithelkarzinom der Haut, ii) asymptomatischem Prostatakrebs ohne bekannte Metastasen und ohne Therapiebedarf oder nur mit Hormontherapie und mit normalem prostataspezifischem Antigen seit ≥ 1 Jahr vor der Randomisierung, oder iii) jeder anderen Krebsart, die seit ≥ 5 Jahren in vollständiger Remission ist
n. Sekundäres Malignom, das die Überlebenszeit auf weniger als 6 Monate begrenzt.
o. Drogen- oder Alkoholmissbrauch innerhalb der letzten 6 Monate
p. Patientinnen, die sich nicht an den Plan zur Schwangerschaftsverhütung halten können
q. Schwangere oder stillende Frauen
r. Thiaminspiegel unter der Normgrenze trotz Supplementierung
s. Patienten, die nicht einwilligungsfähig sind, weil sie die Art, Bedeutung und Tragweite der klinischen Prüfung nicht verstehen und daher keinen vernünftigen Willen im Lichte der Tatsachen bilden können [§ 40 Abs. 1 S. 3 Nr. 3a AMG]
Ruxo-BEAT-Studie
Rekrutierungsstopp am 20.08.2020
Ansprechpartner Zentrum Aachen
Klinik für Hämatologie, Onkologie, Hämostaseologie und
Stammzelltransplantation (Med. Klinik IV)
Pauwelsstraße 30
52074 Aachen
| Prof. Dr. Steffen Koschmieder E-Mail: skoschmieder@ukaachen.de Telefon: 02401-80 - 37929 oder -800 | |
| Projektmanagerin | |
| Kim Kricheldorf E-Mail: kkricheldorf@ukaachen.de Telefon 0241 - 80 - 37029 |
Für ET- und PV-Patienten steht derzeit die sog. Ruxo-BEAT-Studie offen (Abb. 2). Leiter der klinischen Prüfung ist Herr Professor Koschmieder, Universitätsklinikum Aachen. In die Studie können ET- und PV-Patienten mit Indikation zur zytoreduktiven Therapie eingebracht werden. Das Ziel der Ruxo-BEAT-Studie ist es, die Machbarkeit, Sicherheit und Wirksamkeit einer Verabreichung von Ruxolitinib gegenüber der aktuell besten verfügbaren Therapie bei Patienten mit Hochrisiko-PV-oder Hochrisiko-ET zu beurteilen. Primärer Endpunkt ist die Rate der vollständigen klinisch-hämatologischen Ansprechrate nach 6 Monaten.
Ruxolitinib wird in der Ruxo-BEAT-Studie oral in einer Dosis von 10 mg zweimal täglich (PV oder ET) verabreicht. Die bestverfügbare Therapie kann alle derzeit verwendeten Behandlungsmöglichkeiten umfassen und unterliegt der Wahl des Prüfers (Monotherapie mit z.B. Hydroxyurea, Anagrelid, Interferon, Busulfan, Immunmodulatoren etc.). Nachdem eine schriftliche Einwilligung eingeholt wurde, werden in Frage kommende Patienten gescreent. Wenn alle Einschlusskriterien und keine Ausschlusskriterien erfüllt sind, werden die Patienten randomisiert und starten ihre Therapie am Tag 1 der Studie für insgesamt zwei aufeinanderfolgende Jahre.
Eine erweiterte Behandlung mit Ruxolitinib über diesen Zeitpunkt hinaus wird für die Patienten, die auf die Therapie ansprechen, mit der dafür zur Verfügung gestellten Studienmedikation möglich sein. Außerdem ist für Patienten, bei denen die bestverfügbare Therapie nicht wirkt, nach 6 Monaten ein Wechsel in den experimentellen Arm möglich.
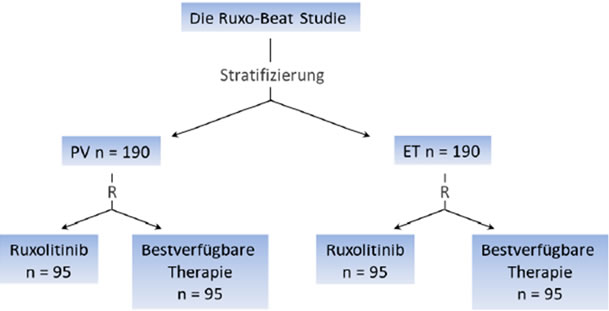
Abb. 2: Übersicht über das Design der Ruxo-BEAT-Studie. Jeweils 190 ET- und PV-Patienten sollen 1:1 randomisiert mit Ruxolitinib bzw. der bestverfügbaren Standardtherapie behandelt werden.
Einschlusskriterien:
- Alter ≥ 60 Jahre
- Vorangegangene Thrombose, Thromboembolie
- Schlechte Verträglichkeit einer Aderlasstherapie
- Häufige Notwendigkeit eines Aderlasses
- Symptomatische oder progressive Vergrößerung der Milz
- Thrombozyten > 1500 • 109 /L
- Schwere krankheitsbedingte Symptome (nach Beurteilung durch den Prüfer)
- Progressive Leukozytose mit einer Leukozytenanzahl >20x109/L
- Alter ≥ 60 Jahre
- Thrombozyten > 1500 • 109 /L
- Vorangegangene Thrombose, Thromboembolie
- Vorangegangene schwerwiegende Blutung aufgrund der ET (definiert als Sinken des Hgb um mindestens 2g/dl)
- Keine vorherige Behandlung mit zytoreduktiven Substanzen mit Ausnahme von Hydroxyurea, Anagrelide oder Interferon für maximal 6 Wochen vor Eintritt in die Studie (Aderlass und Aspirin sind erlaubt)
- Sowohl therapienaive als auch vortherapierte Patienten können eingeschlossen werden.
Ausschlusskriterien:
- Chirurgisch sterilisiert oder mindestens zwei Jahre nach Eintreten der Menopause
- Einwilligung zur Nutzung einer hoch effektiven Verhütungsmethode (Pearl Index < 1), wie orale Kontrazeptive, Intrauterinpessare, sexuelle Abstinenz oder Barrieremethoden (z.B. Kondome) in Kombination mit Spermiziden
POMINC- (MPNSG-0212-) Studie
Rekrutierungsphase abgeschlossen am 27.04.2021
Ansprechpartner Zentrum Ulm
Klinik für Innere Medizin III
Universitätsklinikum Ulm
Albert-Einstein-Allee 23
89081 Ulm
| Prof. Dr. med. Konstanze Döhner E-Mail: konstanze.doehner@uniklinik-ulm.de Telefon 0731 500 45543 Fax 0731 500 45505 |
Prof. Dr. med. Frank Stegelmann E-Mail: frank.stegelmann@uniklinik-ulm.de Telefon 0731 500 45521 Fax 0731 500 45525 |
|
| Studienkoordinatorinnen | ||
| Anke Knödler E-Mail: anke.knoedler @uniklinik-ulm.de Telefon 0731 - 500 - 45919 |
Ulrike Sutter E-Mail: ulrike.sutter @uniklinik-ulm.de Telefon 0731 - 500 - 45929 |
Katrin Vetter E-Mail: katrin.vettter @uniklinik-ulm.de Telefon 0731 - 500 - 45928 |
Bei der POMINC- (MPNSG-0212-) Studie handelt es sich um eine Phase Ib/II Studie zur Kombinationstherapie mit Ruxolitinib und Pomalidomid für Patienten mit primärer und sekundärer MF und Anämie bzw. Transfusionsbedürftigkeit (Abb. 3). Primäre Ziele sind die Evaluation des Behandlungsansprechens von Ruxolitinib und Pomalidomid bei Patienten mit primärer und sekundärer MF unter Verwendung der Konsensuskriterien der Internationalen Arbeitsgruppe für Erforschung und Behandlung von MPN mit Knochenmarkfibrose (IWG-MRT) und dem Kriterium der Transfusionsunabhängigkeit hinsichtlich Erythrozyten und die Evaluation des Sicherheitsprofils der Kombinationstherapie mit Ruxolitinib und Pomalidomid bei Patienten mit primärer und sekundärer MF. Alle Patienten erhalten Ruxolitinib (beginnend mit 2x10 mg tgl., Dosismodifikation möglich) und Pomalidomid (0,5 mg tgl., fixe Dosierung). Gründe bzw. Zeitpunkte für Dosismodifikationen von Ruxolitinib und eine eventuelle Pomalidomid-Therapiepause sind im Protokoll anhand von Verträglichkeit und Ansprechen der Kombinationstherapie definiert. Die Therapie wird, sofern die Erkrankung nicht progredient ist, für mindestens 12 Therapiezyklen (je 28 Tage) durchgeführt. Im Falle eines Ansprechens kann die Behandlung mit den Studienmedikamenten über 12 Zyklen hinaus auf der Grundlage einer individuellen Fallentscheidung erfolgen.
In der Auswertung unserer abgeschlossenen MPNSG-0109-Studie, in der eine Monotherapie mit Pomalidomid bei MF-Patienten mit Anämie und/oder Thrombopenie durchgeführt worden ist, zeigte sich, dass 2 mg Pomalidomid tgl. einer Dosierung von 0,5 mg Pomalidomid tgl. hinsichtlich der Wirksamkeit bei ähnlich guter Verträglichkeit signifikant überlegen ist (Schlenk et al., ASH Annual Meeting Abstract 2013, #2822). Daher ist für die zweite Studienphase (ab Patient 38) der aktuell rekrutierenden POMINC-Studie eine Dosiserhöhung von Pomalidomid von 0,5 mg auf 2 mg tgl. geplant. Zudem hatte sich in der ersten Zwischenauswertung der POMINC-Studie mit 28 eingeschlossenen und behandelten Patienten neben einer unbedenklichen Verträglichkeit der Kombinationstherapie ein vergleichsweise hoher Anteil von etwa 25% der Studienpatienten gezeigt hat, der eine stabile Erkrankung über 12 Zyklen Studientherapie hinaus aufweist und hinsichtlich einer Verlängerung des Transfusionsintervalls bzw. durch einen Rückgang krankheitsassoziierter Symptome von der Kombinationstherapie profitiert (Stegelmann et al., ASH Annual Meeting Abstract 2015, #826). Dies ist insbesondere dahingehend von Bedeutung, weil es sich bei den in der POMINC-Studie behandelten Patienten um ein stark vorbehandeltes MF-Kollektiv mit sehr ungünstiger Prognose handelt (überwiegend Intermediate-II- und Hochrisiko-Patienten nach DIPSS-Stratifizierung [Dynamic International Prognostic Scoring System]).

Abb. 3: Übersicht über das Design und den Ablauf der einarmigen POMINC- (MPNSG-0212-) Studie über 12 Zyklen oder mehr mit Ruxolitinib und Pomalidomid.
Einschlusskriterien:
Ausschlusskriterien:
RuxoAllo-Studie
Rekrutierungsphase abgeschlossen am 31.05.2021
Ansprechpartner Zentrum Hamburg-Eppendorf
Uniklinik Hamburg EppendorfInterdisziplinäre Klinik und Poliklinik für Stammzelltransplantation
Martinistr 52
20246 Hamburg
| Prof. Dr. Nikolaus Kröger E-Mail: Telefon: 040 741054188 |
| Studienkoordination |
| Dr. rer. nat. Liliane Henkes Clinical Research Associate E-Mail: l.henkes@ctc-north.com |
Die dritte IIT der GSG-MPN ist die sog. RuxoAllo-Studie (Abb. 4). Leiter der klinischen Prüfung ist Herr Prof. Nikolaus Kröger, Universitätsklinikum Hamburg-Eppendorf. Hierbei handelt es sich um eine multizentrische Phase-II Studie, die das Überleben von MF-Patienten, die sich aufgrund eines vorhandenen Spenders einer allogenen Blutstammzelltransplantation unterzogen haben, mit dem Überleben von MF-Patienten vergleicht, die keinen passenden Spender haben und kontinuierlich mit Ruxolitinib behandelt werden. Sekundäre Studienziele umfassen den Vergleich von Sicherheit und Wirksamkeit der Behandlungsmodalitäten in den beiden Studienarmen in Bezug auf Milzgrößenreduktion, Verbesserung konstitutioneller Symptome, Lebensqualität, Rückgang der Knochenmarkfibrose und erkrankungsassoziierte Mortalität. Es sollen insgesamt 155 Patienten in einen der beiden Studienarme rekrutiert werden (n=93 allogene Transplantation und n=62 kontinuierliche Ruxolitinib-Therapie).
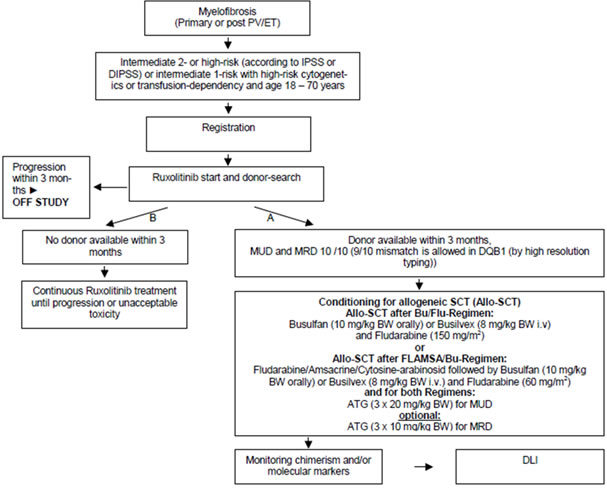
Abb. 4: Übersicht über das Design der RuxoAllo-Studie. Insgesamt 155 Patienten sollen auf die Behandlungsarme A (n=93) und B (n=62) verteilt werden.
Einschlusskriterien:
Ausschlusskriterien:
- Gesamtbilirubin, SGPT oder SGOT > 3-fach über den oberen Normwert erhöht
- Linksventrikuläre Ejektionsfraktion < 30 %
- Kreatinin Clearance < 30 ml/min
- DLCO < 35 % und/oder kontinuierlicher zusätzlicher Oxygenierungsbedarf