Information für Patienten:
Was sind Myeloproliferative Neoplasien?
Definition
Unter dem Begriff „Myeloproliferative Neoplasien“ (MPN) werden mehrere chronische Bluterkrankungen zusammengefasst, deren Ursprung in den sog. Stammzellen liegt. Stammzellen sind im Knochenmark angesiedelt und verantwortlich für die Ausreifung der unterschiedlichen Blutzellen. Bei MPN findet eine überschießende, unkontrollierte Bildung einer oder mehrerer Unterformen von Blutzellen statt. Die Unterteilung erfolgt anhand einer Klassifikation der Weltgesundheitsorganisation (WHO = World Health Organization) (Abb. 1). Die häufigsten Unterformen von MPN sind die Essentielle Thrombozythämie (ET), die Polyzythämia Vera (PV) und die Primäre Myelofibrose (PMF). Vergleichsweise selten sind dagegen beispielsweise die chronische Eosinophilenleukämie (CEL) und die Mastozytose. In manchen Fällen ist eine genaue Klassifizierung durch Überschneidungsformen auch nicht möglich (sog. unklassifizierbare MPN).
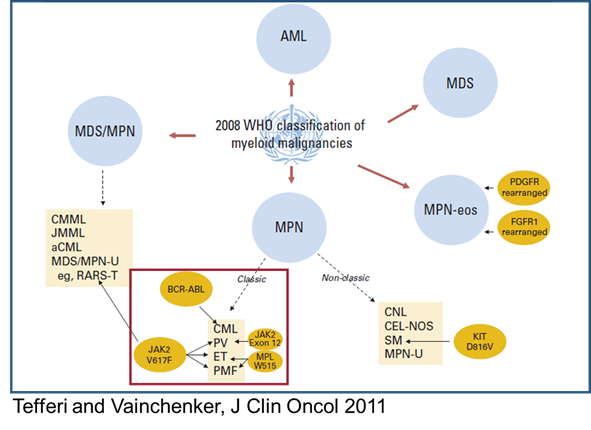
Abb. 1: Klassifikation der myeloischen Neoplasien nach WHO 2007. Die häufigsten (sog. klassischen) myeloproliferativen Neoplasien (MPN) sind rot hervorgehoben (ET, essentielle Thrombozythämie; PV, Polyzythämia vera; PMF, primäre Myelofibrose; CML, chronische myeloische Leukämie).
Wichtig ist die Abgrenzung von der chronischen myeloischen Leukämie (CML), die durch das „Philadelphia-Chromosom“ charakterisiert ist (sog. BCR-ABL-positive CML). Zur Behandlung der CML stehen mittlerweile Tabletten (sog. Tyrosinkinaseinhibitoren) zur Verfügung, die die Leukämiezellen sehr zielgerichtet und nebenwirkungsarm behandeln. Aufgrund ihrer besonderen genetischen und therapeutischen Merkmale wird die CML in dieser Registerstudie nicht berücksichtigt. Die übrigen MPN-Unterformen können daher auch unter dem Begriff BCR-ABL-negative myeloische Neoplasien zusammengefasst werden. Leitbefunde von ET und PV sind eine Vermehrung von Blutplättchen (bei der ET) bzw. roten Blutkörperchen (bei der PV) (Abb. 2). Häufig fallen diese Veränderungen im Rahmen von Routineuntersuchungen auf und verursachen keine Symptome. Manchmal liegen Durchblutungsstörungen kleiner Blutgefäße vor, die Kribbeln in Fingern und/oder Zehen, Kopfschmerzen, Ohrensausen, Augenflimmern oder Juckreiz der Haut verursachen. Ernsthafte Komplikationen sind Verstopfungen von Blutgefäßen durch Blutgerinnsel (sog. Thrombosen oder Embolien) oder - seltener – Blutungen, dies insbesondere in Verbindung mit sehr hohen Blutplättchen-Werten.
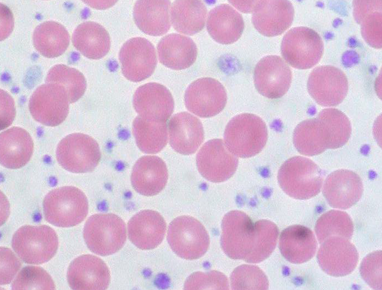
Abb. 2: Blutausstrich von einem Patienten mit ET mit erheblicher Vermehrung der Blutplättchen (lila gefärbt) im Verhältnis zu den deutlich größeren roten Blutkörperchen.
PMF-Patienten sind dagegen - vor allem in der späteren Erkrankungsphase - durch eine Verminderung von Blutzellen (z.B. Anämie = Blutarmut) gekennzeichnet. Diese kommt durch eine langsam voranschreitende Vermehrung von Bindegewebe im Knochenmark (Myelofibrose) mit Verdrängung der normalen Blutbildung zustande (Abb. 3). Leber und Milz, Organe im rechten bzw. linken Oberbauch, können als Reaktion auf die verdrängte Blutbildung beträchtlich anschwellen (sog. Hepatosplenomegalie) und dadurch ein Druckgefühl verursachen.
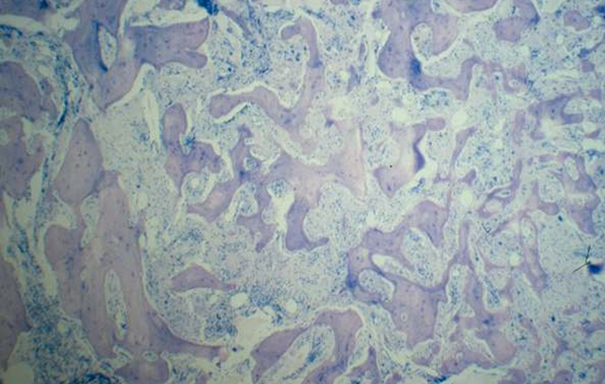
Abb. 3: Knochenmark-Histologie eines Patienten mit fortgeschrittener Myelofibrose. Es zeigt sich eine Vermehrung von Knochenbälkchen (rosa gefärbt, sog. Osteosklerose) mit gleichzeitiger Verdrängung der blutbildenden Areale durch Ausbreitung von Bindegewebe zwischen den Knochenbälkchen.
Prognose
Die Prognose von ET-Patienten ist nach derzeitigem Kenntnisstand trotz der möglichen Gefäßkomplikationen sehr gut. Die Lebenserwartung ist nur in Ausnahmefällen durch Komplikationen der Erkrankung eingeschränkt. Übergänge in eine Myelofibrose sind selten bzw. werden meist erst nach sehr langer Krankheitsdauer (>10 Jahre) beobachtet. Auch die PV gilt insgesamt als prognostisch günstig, während hingegen die Lebenserwartung von PMF-Patienten häufig eingeschränkt ist. Besonders gefürchtet ist dabei der Übergang in eine akute myeloische Leukämie, die häufig schwierig behandelbar ist und daher mit einer schlechten Prognose einhergeht.
Therapie
Die Therapie von MPN reicht daher von alleiniger Beobachtung bis hin zu intensiven Maßnahmen wie der Blutstammzelltransplantation vom Familien- oder Fremdspender (sog. allogene Transplantation), die vor allem für PMF-Patienten mit bestimmten Risikomerkmalen mit dem Ziel der Heilung in Betracht kommt, aber erhebliche Nebenwirkungen bzw. Komplikationen verursachen kann. Bei ET- und PV-Patienten kennt man ebenfalls Risikomerkmale, die eine medikamentöse Therapie notwendig machen. Das Ziel dabei ist allerdings nicht eine Heilung, sondern eine Kontrolle der zu hohen Blutwerte, um vor allem das Risiko für Gefäßkomplikationen (Thrombosen, Embolien, Blutungen) zu senken. Medikamente, die hier zum Einsatz kommen, sind beispielsweise ASS, Hydroxyurea, Anagrelid oder Interferon-alpha. Bei PMF-Patienten ist die Behandlung dagegen oft schwieriger, da die therapeutischen Möglichkeiten zur Verbesserung der Blutbildung begrenzt sind. In dieser Situation kommen neben Bluttransfusionen auch Medikamente in Betracht, die im Rahmen von klinischen Studien getestet werden (z.B. immunmodulatorische Medikamente wie Pomalidomid) und bei einer Subgruppe von Patienten die Blutbildung bessern.
Diagnose
Zur Diagnosestellung sind bei MPN genetische Untersuchungen, also Analysen der Erbsubstanz der Blutzellen von besonderer Bedeutung. Mit den heute verfügbaren Labormethoden kann man genetische Auffälligkeiten in den betroffenen Blutzellen bei etwa 80-90% der Patienten feststellen. Am häufigsten ist eine umschriebene Veränderung der Erbsubstanz (sog. Mutation) in dem Gen Januskinase-2 (JAK2), die sog. JAK2-Mutation. Diese ist bei etwa der Hälfte der ET- und MF- und bei nahezu allen PV-Patienten nachweisbar. Die Mutation spielt in der Erkrankungsentstehung wahrscheinlich eine bedeutende Rolle. Weitere häufige Genmutationen sind die sog. MPL-Mutation und Mutationen im sog. Calreticulin- (CALR-) Gen. Diese drei Mutationen können nach einer Blutentnahme im Labor getestet werden. Eine Knochenmarkuntersuchung ist dafür nicht erforderlich.
Forschung
Im Rahmen wissenschaftlicher Untersuchungen kann zur Klärung der Frage, ob bestimmte genetische Veränderungen spezifisch für die Bluterkrankung sind oder auch im gesunden Körpergewebe vorkommen, die Analyse von nicht von der Bluterkrankung betroffenen Zellen notwendig sein. Als gesundes Körpergewebe kommt hierfür z.B. ein einfach durchzuführender Wangenschleimhautabstrich in Frage, für speziellere Fragestellungen und in selteneren Fällen auch eine kleine, wenige Millimeter große Hautbiopsie, die nach entsprechender lokaler Betäubung mit einer speziellen Hohlnadel auf einfache Weise entnommen werden kann.
Derzeit wird intensiv an der Entwicklung von Medikamenten gearbeitet, die - ähnlich wie bei der CML - zielgerichtet an den erkrankten Blutzellen angreifen und die Erkrankung zurückdrängen sollen. Solche Medikamente sind vom Prinzip her effektiver als unspezifische Ansätze wie beispielsweise bei Hydroxyurea. Bekanntestes Beispiel für eine zielgerichtete Therapieform bei MPN sind die sog. JAK-Inhibitoren. Diese sind insbesondere dazu geeignet, eine Milzvergrößerung und Krankheitssymptome von PMF- und PV-Patienten wirksam zu behandeln. Nachteil ist häufig, vor allem am Anfang der Therapie, ein begleitender Abfall des roten Blutfarbstoffs (Hämoglobin, Hb), das sich in den roten Blutkörperchen befindet und Sauerstoff bindet, und der Blutplättchen. Dabei handelt es sich meist um einen dosisabhängigen Effekt, der am Anfang der Therapie am stärksten ausgeprägt ist. Außerdem kann – insbesondere der Abfall des Hämoglobin-Werts – bei der PV sogar gewünscht sein. Die Entwicklung solcher zielgerichteten Therapieansätze ist jedoch lange noch nicht so weit gediehen wie bei der CML. Dies ist auch daran ablesbar, dass JAK-Inhibitoren bei Patienten ohne JAK2-Mutation wirksam sind und unabhängig von der Mutation eingesetzt werden. Aufgrund der komplexen genetischen Mechanismen gelingt es mit dem derzeitig verfügbaren JAK-Inhibitor Ruxolitinib bislang jedoch noch nicht, die Folgen der Erkrankung wie z.B. die Knochenmarkfibrose effektiv zurückzudrängen.
Die GSG-MPN prüft derzeit Ruxolitinib in drei unterschiedlichen Studien: Zum einen bei ET- und PV-Patienten im Vergleich mit der verfügbaren Standardtherapie („Ruxo-BEAT-Studie“), zum anderen vor einer geplanten allogenen Transplantation bei MF-Patienten („Ruxo-Allo-Studie“) und außerdem in Kombination mit Pomalidomid bei MF-Patienten mit Blutarmut, bei denen eine allogenen Transplantation nicht geplant ist („POMINC-Studie“). Die Details zu diesen Studien finden Sie ebenfalls auf dieser Homepage unter dem Link „Studienaktivitäten“ auf der Startseite.