Wissenschaftliche Projekte der GSG-MPN
MPN-Schwerpunkt Aachen
siehe unter:
Klinik für Hämatologie, Onkologie, Hämostaseologie und Stammzelltransplantation
Uniklinik RWTH Aachen
MPN-Schwerpunkt Bonn (Dominik Wolf)
Ansprechpartner:
Prof. Dr. Dominik Wolf
Medizinische Klinik 3
Onkologie, Hämatologie und Rheumatologie
Sigmund-Freud-Str. 25
53127 Bonn
Tel.: +49(0)228-287-17233
Fax: +49(0)228-287-51729
E-Mail: dominik.wolf@ukb.uni-bonn.de
Interessensgebiete:
Wir beschäftigen uns klinisch und experimentell mit 2 Themenkomplexen im Bereich Philadelphia-negativer MPN:
1. Mechanismen der Wirkung und Nebenwirkungen von JAK-Hemmern in der Therapie von MPNs. Hier gilt unser Hauptaugenmerk den immunologischen Mechanismen der anti-inflammatorischen/immunsupppressiven Effekte der Substanzgruppe. Wir konnten zeigen, daß JAK1/2 Hemmer sehr potent DCs, T Zellen und NK Zellen in Ihrer Funktion hemmen. Die erklärt zum einen die anti-inflammatorische Wirkung (inklusiver möglicher neuer Therapie-Indikationen), aber auch potentielle Nebenwirkungen (Infektionen) dieser Substanzgruppe. Zudem erforschen wir die intrinsische Immundysregulation bei MPNs ohne Therapie, um den Zusammenhang zwischen Inflammation und Symptomen sowie dem klonalen Wachstum besser verstehen zu können.
2. Die Pathogenese splanchnischer Venenthrombosen auf der Basis einer MPN ist unser zweites Interessensgebiet. In enger Zusammenarbeit mit Prof. Trebicka (Medizinische Klinik 1/UKB Bonn), der Transfusionsmedizin (Prof. Oldenburg und Prof. Pötzsch/UKB Bonn) und Partnern aus der GSG-MPN (Prof. Koschmieder/UK Aachen und Prof. Grieshammer/Klinikum Minden) werden potentielle Risikofaktoren identifiziert und Pathomechanismen dieser klinisch relevanten vaskulären Komplikation untersucht. Ziel ist über ein verbessertes Verständnis der Pathogenese und Definition individueller Risikofaktoren in Zukunft Patienten bereits vor Auftreten des Gefäßereignisses identifizieren und (zB über eine Blutverdünnung) besser schützen zu können.
MPN-Schwerpunkt Essen (J. Göthert)
Ansprechpartner:
Dr. Joachim R. Göthert
Universitätsklinikum Essen
Klinik für Hämatologie
Westdeutsches Tumorzentrum (WTZ)
Email joachim.goethert@uk-essen.de
Tel.: 0201-723-5136
Fax: 0201-723-5934
Interessensgebiete:
Der Schwerpunkt myeloproliferative Neoplasien ist daran interessiert, die medizinische Versorgung und spezifische Behandlung von MPN-Patienten zu verbessern. Im Zentrum steht hierbei die Teilnahme an der Registerstudie der GSG-MPN, um die Dokumentation klinischer Verläufe zu dokumentieren und Biomaterial zu asservieren. Im Speziellen nimmt der MPN-Schwerpunkt Essen an mehreren multizentrischen Studien teil. Diese umfassen unter anderem die ReTHINK Studie, die den Einsatz von Ruxolitinib bei Patienten mit Hochrisiko-Mutationen im Frühstadium der primären Myelofibrose prüft. Weiterhin nimmt der Essener Schwerpunkt an der Ruxo-BEAT Studie teil, die die Behandlung mit Ruxolitinib gegen die beste verfügbare Therapie bei Hochrisiko Polycythaemia vera und essentieller Thrombozythämie prüft. Weitere Studienteilnahmen befinden sich in der Vorbereitungsphase. Ferner kann Patienten mit einer aggressiven Mastozytose über ein Härtefallprogramm die Behandlung mit dem Multikinaseinhibitor Midostaurin ermöglicht werden. Weitere Forschungsinteressen des GSG-Standortes Essen umfassen die Veränderungen von neutrophilen Granulozyten bei MPN, Wirkmechanismen von JAK-Kinaseninhibitoren, splanchnische Thrombosen bei MPN und in vivo Modelle myeloischer Neoplasien.
MPN-Schwerpunkt Freiburg
Ansprechpartner:
Prof. Dr. Nikolas von Bubnoff
Klinik für Innere Medizin I
Klinik für Tumorbiologie
Schwerpunkt Hämatologie, Onkologie und Stammzelltransplantation
Universitätsklinikum Freiburg
Hugstetter Str. 55
79106 Freiburg
Tel: +49-761-270-33210
Fax: +49-761-270-73570
Email: nikolas.bubnoff@uniklinik-freiburg.de
Prof. Dr. Heike L. Pahl
Klinik für Innere Medizin I
Klinik für Tumorbiologie
Schwerpunkt Hämatologie, Onkologie und Stammzelltransplantation
Universitätsklinikum Freiburg
Hugstetter Str. 55
79106 Freiburg
Tel: +49-761-270-33210
Fax: +49-761-270-73570
Email: nikolas.bubnoff@uniklinik-freiburg.de
Interessensgebiete / Projekte
A) Behandlung der GvHD mit Ruxolitinib
- Auch MPN Patienten entwickeln häufig nach KMT eine GvHD
- Pilotstudie an Patienten mit steroid-refraktärer GvHD (alle Grunderkrankungen, nicht nur MPN Patienten)
- Ansprechrate:
- akute GvHD :81%, 46% komplette Remission
- chronische GvHD: 85 %
- 6 Monats-Überlebensrate
- akute GvHD: 79%
- chronische GvHD: 97 %
- Ansprechrate:
- Multizentrische Phase II Studie, Beginn 4. Quartal 2016
B) Identifikation und Charakterisierung neuer therapeutischer Zielstrukturen bei MPN
- Sequenzierung von Proben informativer Patienten
- Untersuchung von Resistenz-Mechanismen
- Simulation Mutations-Unabhängiger Resistenz in vitro und in vivo
- Charakterisierung betroffener Signaltransduktionswege
C) Molekulare Pathogenese der MPN
- Beschreibung neuer molekularer Marker
- Identifikation neuer Mutationen bzw. aberranter Signalwege ursächlich für die Entstehung oder Progression von MPN
D) Murine Modelle der MPN
- Etablierung und Charakterisierung muriner MPN Modelle
- NF-E2: Einziges Maus-Modell, welches die spontane Transformation in eine akute Leukämie zeigt
- Untersuchung der Pathomechanismen leukämischer Transformation bei MPN anhand dieses Modells
E) Freiburger MPN Register
- Etablierte lokale klinische Datenbank und Biobank zur Unterstützung der translationalen/experimentellen Erforschung der MPN
Ausgewählte Publikationen:
- Gorantla SP, Zirlik K, Reiter A, Yu C, Illert AL, von Bubnoff N*, Duyster J* (2015). F604S exchange in FIP1L1-PDGFRA enhances FIP1L1-PDGFRA protein stability via SHP-2 and SRC: a novel mode of kinase inhibitor resistance. Leukemia 29(8):1763-70. * contributed equally.
- Spoerl S, Mathew NR, Bscheider M, Schmitt-Graeff A, Chen S, Mueller T, Verbeek M, Fischer J, Otten V, Schmickl M, Maas-Bauer K, Finke J, Peschel C, Duyster J, Poeck H, Zeiser R, von Bubnoff N (2014). Activity of therapeutic JAK 1/2 blockade in graft-versus-host disease. Blood 12: 3832-42.
- von Bubnoff N, Gorantla SP, Engh RA, de Oliveira T, Thöne S, Aberg E, Peschel C, Duyster J (2011). The low frequency of clinical resistance to PDGFR inhibitors in myeloid neoplasms with abnormalities of PDGFRA might be related to the limited repertoire of possible PDGFRA kinase domain mutations in vitro. Oncogene 30: 933-43.
- von Bubnoff N, Engh RA, Aberg E, Sanger J, Peschel C, and Duyster J. (2009). FMS-like tyrosine kinase 3-internal tandem duplication tyrosine kinase inhibitors display a nonoverlapping profile of resistance mutations in vitro. Cancer Res 69: 3032-3041.
- von Bubnoff N, Barwisch S, Speicher MR, Peschel C, Duyster J (2005). A cell-based screening strategy that predicts mutations in oncogenic tyrosine kinases: implications for clinical resistance in targeted cancer treatment. Cell Cycle 4:400-406.
- Kaufmann KB, Gründer A, Hadlich T, Wehrle J, Gothwal M, Bogeska R, Seeger TS, Kayser S, Pham KB, Jutzi JS, Ganzenmüller L, Steinemann D, Schlegelberger B, Wagner JM, Jung M, Will B, Steidl U, Aumann K, Werner M, Günther T, Schüle R, Rambaldi A, Pahl HL, (2012) A novel murine model of myeloproliferative disorders generated by overexpressionof the transcription factor NF-E2. J. Exp. Med, 209: 35 - 50.
- Jutzi JS, Bogeska R, Nikoloski G, Schmid CA, Seeger TS, Stegelmann F, Schwemmers S, Gründer A, Peeken J, Gothwal M, Wehrle J, Aumann K, Hamdi K, Dierks, C, Wang W, Döhner K, Jansen JH, Pahl HL (2013) MPN patients harbor recurrent truncating mutations in transcription factor NF-E2, J. Ex. Med. 210: 1003 - 1019.
- Aumann K, Frey AV, May AM, Hauschke D, Kreutz C, Marx JP, Timmer J, Werner M, Pahl HL (2013) Subcellular mislocalization of the transcription factor NF-E2 in erythroid cells discriminates pre-fibrotic primary myelofibrosis from essential thrombocythemia. Blood 122: 93 - 99.
- Hexner EO, Mascarenhas J, Prchal J, Roboz GJ, Baer MR, Ritchie EK, Leibowitz D, Demakos EP, Miller C, Siuty J, Kleczko J, Price L, Jeschke G, Weinberg R, Basu T T, Pahl HL, Orazi A, Najfeld V, Marchioli R, Goldberg JD, Silverman LR, Hoffman R. (2015) Phase I dose escalation study of lestaurtinib in patients with myelofibrosis. Leuk. Lymph. 7: 1 – 26.
- Geyer H, Scherber R, Kosiorek H, Dueck AC, Kiladjian JJ, Xiao Z, Slot S, Zweegman S, Sackmann F, Fuentes AK, Hernández-Maraver D, Döhner K, Harrison CN, Radia D, Muxi P, Besses C, Cervantes F, Johansson PL, Andreasson B, Rambaldi A, Barbui T, Bonatz K, Reiter A, Boyer F, Etienne G, Ianotto JC, Ranta D, Roy L, Cahn JY, Maldonado N, Barosi G, Ferrari ML, Gale RP, Birgegard G, Xu Z, Zhang Y, Sun X, Xu J, Zhang P, te Boekhorst PAW, Commandeur S, Schouten H, Pahl HL, Griesshammer M, Stegelmann F, Lehmann T, Senyak Z, Vannucchi AM, Passamonti F, Samuelsson J, Mesa RA (2016) Symptomatic Profiles of Patients with Polycythemia Vera: Implications of Inadequately Controlled Disease, J. Clin. Oncol. 34: 151-159.
- Klein C, Zwick A, Kissel S, Forster CU, Pfeifer D, Follo M, Illert AL, Decker S, Benkler T, Pahl HL, Oostendorp RAJ, Aumann K, Duyster J, Dierks C (2016) Ptch2 loss drives myeloproliferation and myeloproliferative neoplasm progression, J. Exp. Med. 213: 273-290.
MPN-Schwerpunkt Hamburg-Eppendorf (N. Kröger)
Ansprechpartner:
Prof. Dr. N. Kröger, Dr. I. Triviai, PD Dr. M. Christopeit, C. Stocking, A. Badbaran, S. Zeschke,
T. Stübig, Prof. Dr. B. Fehse, T. Derlin, Dipl.-Biol. T Stahl
Interdisziplinäre Klinik und Poliklinik für Stammzelltransplantation
Onkologisches Zentrum
Martinistr. 52
20246 Hamburg
Tel.: +49 (0) 40-7410-55864; +49 (0) 40-7410-54851 (Sekr.); +49 (0) 40-7410-55250 (Sekr.)
Fax: +49 (0) 40-7410-53795
E-Mail: nkroeger@uke.de
Interessensgebiete:
1. Entwicklung allogener Transplantationskonzepte bei Myelofibrose 2. Optimierung des molekularen Monitoring zur Detektion von minimaler Restkrankheit (qPCR, NGS, dPCR) 3. Entwicklung eines Xenotransplantationsmodels für Myelofibrose 4. Charakterisierung der Myelofibrose-„Stammzelle“ und deren Interaktion mit Mikroenvironement 5. Molekulare Regulationsmechanismen in der Entwicklung der Knochenmarkfibrose 6. Immunologisches Monitoring unter JAK 1/2 Inhibitoren 7. Evaluation von PET-CT in Myelofibrose
Drittmittelförderungen durch:
- Deutsche Krebshilfe
- European Hematology Association (EHA)
- Hamburger Krebsgesellschaft
- Erich & Gertrud Roggenbuck Stiftung
Ausgewählte Publikationen:
1. Triviai I, et al.: CD133 marks marks a stem cell population that drives human primary myelofibrosis. Haema-tologica. 2015 Jun;100(6):768-79 2. Triviai I, et al.. Endogenous retrovirus induces leukemia in a xenograft mouse model for primary myelofi-brosis. Pro Natl Acad Sci USA. 2014 Jun 10;111(23):8595-600 3. Kröger N, et al.: Rapid regression of bone marrow fibrosis after dose-reduced allogeneic stem cell trans-plantation in patients with primary myelofibrosis. MDS-Subcommittee of the Chronic Leukeamia Working Party of the European Group for Blood and Marrow Transplantation. Exp Hematol. 2007 Nov;35(11):1719-22 4. Kröger N, et al.: JAK2-V617F-triggered preemptive and salvage adoptive immunotherapy with donor-lymphocyte infusion in Patients with myelofibrosis after allogeneic stem cell transplantation. Blood. 2009 Feb 19;113(8):1866-8 5. Kröger N, et al.: Allogeneic stem cell transplantation after reduced-intensity conditioning in patients with myelofibrosis: a prospective, multicenter study of the Chronic Leukemia Working Party of the European Group for Blood and Marrow Transplantation. Blood. 2009 Dec 17;114(36):5246-70 6. Alchalby H, et al.: Impact of JAK2V617F mutation status, allele burden, and clearance after allogeneic stem cell transplantation for myelofibrosis. Blood. 2010 Nov 4;116(18):3572-81 7. Kröger N, et al.: Impact of allogeneic stem cell transplantation on survival of patients less than 65 years of age with primary myelofibrosis. Blood. 2015 May 21;125(21):3347-50 8. Stübig T et al.: JAK inhibition with ruxolitinib as pretreatment for allogeneic stem cell transplantation in primary or post-ET/PV myelofibrosis. Leukemia. 2014 8;28(8):1736-8 9. Derlin T, et. al.: Assessment of bone marrow inflammation in patients with myelofibrosis: an 18F-fluorodeoxyglucose PET/CT study. Eur J Nucl Med Mol Imaging. 2015 4;42(5):696-705.
MPN-Schwerpunkt Jena (Florian Heidel, Thomas Ernst und Andreas Hochhaus )
Ansprechpartner:
Prof. Dr. Florian Heidel, PD Dr Thomas Ernst, Prof. Dr. Andreas Hochhaus
Universitätsklinikum Jena
Klinik für Innere Medizin II
Abteilung Hämatologie und Internistische Onkologie
Erlanger Allee 101
07747 Jena
Tel.: +49 3641 9-324201
Fax: +49 3641 9-324202
E-Mail: onkologie@med.uni-jena.de
Interessensgebiete:
Identifikation und funktionelle Charakterisierung von Zielstrukturen zur Eradikation von JAK2V617F mutierten Stamm- und Progenitorzellen (Prof. Dr. Florian Heidel, Universitätsklinikum Jena)
Philadelphia-Chromosom negative myeloproliferative Neoplasien (MPN) sind klonale Erkrankungen alternder hämatopoetischer Stammzellen sowie früher myeloischer Vorläuferzellen.
MPNs mit einer JAK2V617F-Mutation zeichnen sich durch eine vermehrte Proliferation einer oder mehrerer myeloischer Zellreihen aus sowie durch eine ausgeprägte systemische Entzündungsreaktion, vor allem in fortgeschrittenen Stadien der Erkrankung.
Die molekularen Mechanismen, die der JAK2V617F-induzierten Pathogenese zugrunde liegen, sind weitgehend unbekannt. Die therapeutischen Maßnahmen bei JAK2V617F-positiven MPNs sind bis dato auf eine symptomatische Behandlung der Patienten limitiert. Ziel unserer Gruppe ist es, die molekularen Pathomechanismen dieser Erkrankung zu identifizieren und zu verstehen. Mittels molekularen Screening-Untersuchungen (RNAi-basiert oder CRISPR/Cas9-basiert) identifizieren wir potentielle neue molekulare Zielstrukturen in JAK2V617F mutierten Stamm- und Progenitorzellen. Zudem führen wir funktionelle Untersuchungen in vitro und in vivo durch, um Zielstrukturen zu charakterisieren, welche zur Entstehung und Erhaltung der Erkrankung beitragen. Die Untersuchungen werden hier sowohl an murinen als auch an humanen Stamm- und Progenitorzellen durchgeführt und funktionell in Mausmodellen charakterisiert.
Dysregulation der Integrin-Funktion und Induktion der Entzündung in JAK2V617F mutierten Myeloproliferativen Neoplasien (Prof. Dr. Florian Heidel, Universitätsklinikum Jena, gemeinsam mit der Gruppe von Prof. Dr. Th. Fischer, OvGU Magdeburg)
Molekularbiologische, tierexperimentelle und klinische Daten zeigen eine ausgeprägte Zytokinhypersensitivität der neoplastischen hämatopoetischen Progenitorzellen bei JAK2V617F mutierten Myeloproliferativen Neoplasien (MPN). Charakteristisch ist zudem die Induktion einer ausgeprägten systemischen Entzündungsreaktion in fortgeschrittenen Phasen der Erkrankung. Beide Phänomene spielen eine entscheidende Rolle für den klinisch sichtbaren Phänotyp. Die molekularen Mechanismen, die der JAK2V617F-induzierten Pathogenese und insbesondere der systemischen Entzündungsreaktion zugrunde liegen, sind weitgehend unbekannt und sollen in diesem Projekt molekularbiologisch charakterisiert werden. Im Fokus des Projektes steht der PLCg1-Signalweg, den wir in unserer letzten Förderperiode als wichtigen Regulator der terminalen erythropoetischen Differenzierung charakterisieren konnten. Der Einfluss von PLCg1 auf die mit der myeloproliferativen Neoplasie vergesellschafteten Entzündungsreaktion und ihre Folgen wird in der aktuellen Förderperiode in vitro und in vivo untersucht.
Förderung: Deutsche Forschungsgemeinschaft (DFG); Sonderforschungsbereich 854 (SFB854, Teilprojekt A20).
Klonale Evolution in MDS/MPN-Erkrankungen (PD Dr. Thomas Ernst, Universitätsklinikum Jena)
Durch den Einsatz moderner Technologien konnten in den vergangenen Jahren zahlreiche neue molekulare Aberrationen in Patienten mit MDS/MPN-Erkrankungen (z.B. atypische CML, CMML) aufgedeckt werden, welche eine zunehmende Bedeutung zur Differenzialdiagnostik, Prognoseabschätzung, Resistenzaufklärung und Therapiewahl erlangen. In einem aktuellen Projekt der Arbeitsgruppe wurde eine auf der 454-Technologie basierende Next-Generation-Sequencing-Methode zum sensitiven und simultanen Nachweis der 30 häufigsten Leukämie-assoziierten Gene in einem einzigen Lauf etabliert („Jena-454-Leukämie-Panel“). Mittels ultra-tiefer Sequenzierung können nachgewiesene Patienten-spezifische Mutationen im Anschluss an die Erstdiagnose im Krankheitsverlauf unter konventionell medikamentöser Therapie oder nach allogener Stammzelltransplantation mit sehr hoher Sensitivität gezielt nachkontrolliert und beobachtet werden. Das Verständnis, wie genetische und epigenetische Veränderungen zusammenpassen, deren klinische Bedeutung, die klonale Hierarchie, die Kinetik einzelner und zusammenwirkender Mutationen und wie diese Veränderungen letztendlich effektiv durch zielgerichtete Therapien angegangen werden können, könnten in Zukunft die Behandlung von Patienten mit MDS/MPN-Erkrankungen verbessern.
MPN-Schwerpunkt Magdeburg (Thomas Fischer)
Ansprechpartner:
Prof. Dr. Thomas Fischer
Universitätsklinikum Magdeburg
Direktor der Klinik für Hämatologie und Onkologie
Leipziger Str. 44
39120 Magdeburg
Tel.: +49 391 67-13266
Fax: +49 391 67-13267
E-Mail: thomas.fischer@med.ovgu.de
Interessensgebiete:
Bei den chronisch myeloproliferativen Neoplasien wird im Rahmen des Sonderforschungsbereichs SFB-854 die Rolle der JAK2-V617F Mutation bei der Entstehung der Inflammationsreaktion im Patienten untersucht. Diese Forschungsarbeiten werden sowohl in Zelllinien, in Mausmodellen als auch in Blutzellen von Patienten durchgeführt.
Molekularbiologische, tierexperimentelle und klinische Daten zeigen eine ausgeprägte Zytokinhypersensitivität der neoplastischen hämatopoetischen Progenitorzellen und die Induktion einer systemischen Entzündungsreaktion in fortgeschrittenen Phasen der Erkrankung. Beide Phänomene spielen eine entscheidende Rolle für den klinisch sichtbaren Phänotyp. Die molekularen Mechanismen, die der JAK2V617F-induzierten Pathogenese und der systemischen Entzündungsreaktion zugrunde liegen, sind weitgehend unbekannt und sollen in diesem Projekt molekularbiologisch charakterisiert werden. Dabei steht der Btk/PLC-γ1 Signalweg im Mittelpunkt unseres Interesses (Abb. 1). Der Btk/PLC-γ1 Signalweg ist zentraler Bestandteil eines positiven Rückkopplungsmechanismus bei der Erythropoetinrezeptor-vermittelten Signaltransduktion. Zusammengefasst erwarten wir von unseren Untersuchungen ein tieferes Verständnis zur Bedeutung inflammatorischer Signalwege für die Pathogenese der MPN.
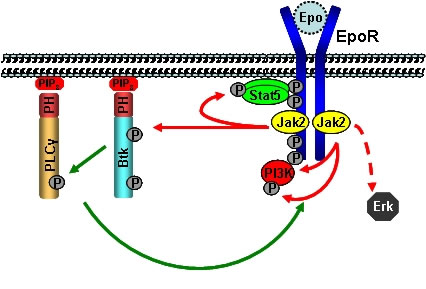
Abbildung 1: Vereinfachte schematische Darstellung der Btk und PLC-γ Signalwege downstream des EpoR/JAK2 Signalkomplexes. Die Stimulation des EpoR mit Erythropoietin führt zur Aktivierung von STAT5, PI3K und ERK. Die Aktivierung der PI3K generiert PIP3, wodurch Btk und PLC-γ über ihre PH-Domänen an die Membran rekrutiert werden. (modifiziert nach von Lindern et al., Cell Cycle 2004).
In der vergangenen Förderperiode des SFB-854 stand dabei die Aufklärung der Rolle des Btk/PLC-γ1 Signalweges für die aberrante zelluläre Zytokinantwort und die Induktion einer systemischen Entzündungsreaktion in JAK2V617F-mutierten myeloproliferativen Neoplasien im Vordergrund. Aktuell wird die aberrante Aktivierung von Integrinen auf Leukozyten untersucht. Innovative Phase I und II Studien mit neuen Signalinhibitoren begleiten diese Forschungsarbeiten auf klinischer Ebene.
Im Rahmen des BMBF-geförderten Verbundprojekts e.bio JAK-Sys „Understanding misbalanced signalling by JAK2-V617F in myloproliferative neoplasms fusing qualitative and quantitative modelling“ wird die molekulare Organisation der JAK2-V617F Signaltransduktion erforscht. Es handelt sich um ein multidisziplinäres Forschungsprojekt der Universität Magdeburg zusammen mit dem Max- Planck- Institut.
MPN-Schwerpunkt Mainz, Thomas Kindler
Ansprechpartner
PD Dr. med. Thomas Kindler
Department of Hematology, Medical Oncololgy & Pneumology
University Medical Center of Mainz
Langenbeckstr. 1, 55101 Mainz
Germany
Phone: 0049-(0)6131-175914
Fax: 0049-(0)6131-17475046
E-Mail: thomas.kindler@unimedizin-mainz.de
Studien:
- Tripple-Kombinationsstudie PIM-LEE-RUXO
- Phase 1b-Studie für Patienten mit Myelofibrose, welche unter einer adäquaten Ruxolitinib-Monotherapie kein zufriedenstellendes Ansprechen erreichen
- Therapie mit Ruxolitinib in Kombination mit dem PIM-Inhibitor PIM447 und dem CDK4/6-Inhibitor LEE011
- POMINC-Studie:
- Phase 1/2-Studie für Patienten mit Myelofibrose and gleichzeitig vorliegender Anämie und Splenomegalie
- Therapie mit Ruxolitinib in Kombination mit Pomalidomid
- ReThink-Studie
- Phase 3-Studie für Patienten mit einer unbehandelten Myelofibrose im Frühstadium und Vorliegen einer Hochrisikomutation
- Therapie mit Ruxolitinib versus Placebo nach Randomisierung
- Ruxolitinib versus allogene Stammzelltransplantation (SZT)
- Phase 2-Studie für Patienten mit einer Indikation zur allogenen SZT
- Therapie mit allogener SZT oder Ruxolitinib in Abh. der Spenderverfügbarkeit
- RuxoBEAT-Studie
- Phase 3-Studie für Patienten mit Polycythämia vera oder Essentieller Thrombozythämie mir vorliegender Indikation zur Einleitung einer zytoreduktiven Therapie
- Therapie mit Ruxolitinib versus der besten zur Verfügung-stehenden Standardtherapie nach Randomisierung
Ansprechpartner:
PD Dr. Thomas Kindler (thomas.kindler@unimedizin-mainz.de)
Website: http://www.unimedizin-mainz.de/uct/studien/klinische-studien/tumortherapie-studien.html
Interessengebiete:
Untersuchung der Thrombozyten und Leukozytenfunktion bei Patienten mit Essentieller Thrombozythämie (ET)Die Essentielle Thrombozythämie (ET) ist eine klonale myeloproliferative Neoplasie (MPN). Trotz einer fast normalen Lebenserwartung ist diese Erkrankung mit einem hohen Risiko von vaskulären thrombotischen Ereignissen, einschließlich ischämischem Schlaganfall, akutem Myokardinfarkt und venöser Thromboembolie, assoziiert. Die verantwortlichen Pathomechanismen, die zu vaskulären Ereignissen bei einzelnen Patienten führen, bei anderen wiederum nicht, sind bisher nur unvollständig verstanden. Auf der Grundlage unserer Hypothese, dass eine Thrombozyten-Dysfunktion eher als die Thrombozyten-Konzentration zu dieser klinischen Heterogenität beiträgt, ist unser Ziel das hämostaserelevante und inflammatorische Thrombozyten-Aktivierungspotenzial von ET-Patienten umfassend zu untersuchen. Patienten mit reaktiver Thrombozytose werden als Kontrollen dienen. Neben klassischen Thrombozyten-Funktionsparametern werden wir das quantitative Phospho- und Thiolredox-Proteom, Zytokin-Expressionsprofil, Leukozytenfunktion und Thrombozyten-Leukozyten-Interaktionen in vitro analysieren. Diese funktionellen Studien werden durch Thrombozyten-Transkriptom-Analysen komplementiert, um zusätzlich relevante genetische Signaturen zu identifizieren. Diese Untersuchungen werden uns helfen, sowohl neue Pathomechanismen in Bezug auf thromboembolische und/oder hämorrhagische Risiken sowie somatische Mutationen zu identifizieren, als auch prädiktive Marker für klinische Manifestationen bei ET-Patienten zu definieren. Die Patienten werden regelmäßig in jährlichen Abständen bis mindestens zum Ende der Studie sowohl klinisch als auch experimentell mit Hilfe von Thrombozyten- und Leukozyten-Funktionsanalysen untersucht. Wir erwarten, dass diese grundlagenforschungsorientierte „proof-of-concept“ Studie die Grundlage für eine groß angelegte Kohortenstudie bildet, um prospektiv individuelle Risikofaktoren bei ET-Patienten zu identifizieren und um auch Behandlungsstrategien zu evaluieren, die vaskuläre Ereignisse und assoziierte Begleiterkrankungen verhindern können.
Förderung: BMBF
Ansprechpartner:
PD Dr. med. Thomas Kindler, III. Medizinische Klinik und Poliklinik, Universitätsmedizin Mainz PD Dr. rer. nat. Kerstin Jurk, Centrum für Thrombose und Hämostase (CTH), Universitätsmedizin MainzPrävalenz MPN-assoziierter Mutationen in Kontext veränderter Blutbildparameter innerhalb einer prospektiven Bevölkerungsstudie
Die Gutenberg-Gesundheitsstudie ist eine groß angelegte, prospektive und repräsentative Bevölkerungsstudie. Im Rahmen des Projektes wird der Gesundheitszustand der Bevölkerung in der Rhein-Main-Region untersucht. In den Jahren 2007 bis 2012 wurden über 15.000 Personen aus einer repräsentativen Bevölkerungsstichprobe in die Studie eingeschlossen. Die Durchsicht der erhobenen Blutbildparameter zeigte, dass ein Teil der Personen veränderte, außerhalb der Norm liegende Werte für Leukozyten, Thrombozyten und Hämatokrit aufwies. Im Rahmen dieses Projektes sollen veränderte Blutbildparameter mit dem Auftreten von vaskulären Ereignissen (arterielle und venöse Thrombosen, Blutungen) assoziiert werden. Darüber hinaus wird das Vorliegen von erworbenen genetischen Veränderungen, assoziiert mit myeloproliferativen Neoplasien (MPN; JAK2V617F, CALR-Mutation, MPLW515-Mutation), untersucht. Ziel der Studie ist es, die Prävalenz von MPNs in einer ansonsten gesunden Bevölkerung zu evaluieren und ggf. Indikatoren zu entwickeln, welche ein frühes Erkennen von Patienten mit erhöhtem vaskulärem Risiko ermöglichen.
Ansprechpartner:
PD Dr. med. Thomas Kindler, III. Medizinische Klinik und Poliklinik, Universitätsmedizin Mainz
Prof. Dr. med. Philipp Wild, Präventive Kardiologie und Medizinische Prävention, Universitätsmedizin Mainz
MPN-Schwerpunkt Minden, Martin Griesshammer
Ansprechpartner
Professor Dr. Martin Griesshammer
Direktor der Universitätsklinik für
Hämatologie, Onkologie,Gerinnungsstörungen und Palliativmedizin
Johannes Wesling Klinikum Minden
UKRUB, Ruhr-Universiät Bochum
Hans-Nolte-Straße 1, D-32429 Minden
Tel.:(+49) 0571-790-4201
E-Mail: martin.griesshammer@muehlenkreiskliniken.de
Interessengebiete
1. European LeukemiaNet Registry: Prospektive und Retrospektive Daten-Erhebung im Rahmen der Beobachtungsstudie des European LeukemiaNet Projektes „Pregnancy in MPN“Aufgrund der Seltenheit und dem bisher nur ungenügend durch Daten untermauerten therapeutischen Vorgehen bei dem Problem Schwangerschaft bei BCR-ABL negativen chronischen myeloproliferativen Erkrankungen wurde innerhalb des European LeukemiaNet Projektes 2006 ein Register zur Dokumentation von Schwangerschaften bei bcr/abl-negativen chronischen myeloproliferativen Erkrankungen implementiert. Die entsprechenden case reports forms (CRFs) und weitere Informationen einschließlich Ethikvotum sind über den e-mail Kontakt mit Prof. Dr. Martin Griesshammer zu erhalten. Auf Anfrage übersendet er auch gerne einen für dieses Projekt erarbeiten Managementvorschlag oder steht für einen konsiliarischen Rat zur Verfügung. In jedem Fall sollte eine Dokumentation über die CRFs erfolgen, so dass zukünftig auf der Basis dieser Erfahrungen ein durch Daten besser abgesichertes und erfolgreicheres Ergebnis erzielt werden kann. Im Anhang der aktuelle Stand des Registers nach Meldung an das Kompetenznetz Leukämien.
2. Retrospektive Erfassung von Hautveränderungen, die durch eine zytoreduktive Behandlung mit Hydroxyurea, Anagrelide oder Interferon alpha bei Patienten mit chonischen myeloproliferativen Neoplasien verursacht wurden, Retrospektive, nicht-interventionelle BeobachtungsstudieRationale
In der Literatur finden sich zahlreiche Hinweise für das Auftreten von Hautveränderungen nach zytoreduktiver Behandlung chronischer myeloproliferativer Neoplasien (MPN). Diese Hautveränderungen wurden insbesondere bei zytoreduktiver Behandlung mit Hydroxyurea beschreiben. Dabei wurden mannigfaltige dermatologische Komplikationen (Ulcera, aktinische Keratosen, vermehrt Plattenepithel-Karzinome, Mundaphthen, Hyperpigmentierungen von Haut und Nägeln, Dermatomyositis-ähnliche und Lichen planus-ähnliche Eruptionen etc.) unter der Behandlung mit Hydroxyurea beobachtet. Diese Hautveränderungen werden dann allerdings häufig eher mit den Komplikationen (wie z.B. Mikrozirkulationsstörungen etc.) bei diesen Erkrankungen in Verbindung gebracht oder gar als völlig unabhängig von diesen Erkrankungen betrachtet. Letztendlich sind aber beispielsweise Hautnekrosen typische Komplikationen der Behandlung mit Hydroxyurea und nur das Absetzen dieser Substanz führt zum Verschwinden der Nekrosen. Andererseits wurden auch unter den anderen eingesetzten zytoreduktiven Medikamenten wie Interferon alpha und Anagrelide pathologische Hautveränderungen beobachtet. Es erscheint allerdings, dass diese im Vergleich zum Hydroxyurea in geringerem Umfang pathologische Hautveränderungen verursachen. Systematische und vergleichende Analysen über das Auftreten von Hautveränderungen nach zytoreduktiver Behandlung chronischer myeloproliferativer Neoplasien sind in der Literatur bisher nicht vorhanden.
Ziel und Umfang der retrospektiven Analyse
Das primäre Ziel dieser retrospektiven Analyse ist die Erfassung der Häufigkeit von dermatologischen Veränderungen durch die zytoreduktive Therapie bei Patienten mit chronischen myeloproliferativen Neoplasien. Folgende chronische myeloproliferative Neoplasien sollen untersucht werden: Essentielle Thrombozythämie (ET), Polycythämia vera (PV) und die primäre Myelofibrose (PMF). Folgende zytoreduktive Medikamente werden in dieser retrospektiven Analyse einbezogen: Hydroxyurea, Interferon alpha und Anagrelide.
Praktische Durchführung und Orte der retrospektiven Analyse
Um die Häufigkeit von Hautveränderungen durch zytoreduktive Medikamente bei Patienten mit chronischen myeloproliferativen Neoplasien (MPN) zu erfassen, erfolgt eine retrospektive Analyse der Patientenakten aus zwei hämatologischen Zentren, die sich schwerpunktmäßig mit diesen Erkrankungen befassen: Das Universitätsklinikum Ulm, Abtleilung Hämatologie und Onkologie, Direktor: Prof. Dr. H. Döhner und die Abtleilung Hämatologie/Onkologie und Palliativmedizin des Klinikums Minden, Direktor: Prof. Dr. M. Grießhammer. Für diese Analyse sollen alle Patienten mit CMPN (ET, PV und PMF), die in den Ambulanzen der beiden Kliniken in den Jahren ab 2010 behandelt wurden, herangezogen werden. Dies dürften nach Schätzungen ca. 200 Patienten sein.
Primärer Endpunkt
Häufigkeit und Art dermatologischer Veränderungen, die durch die zytoreduktive Behandlung mit Hydroxyurea, Anagrelide oder Interferon alpha aufgetreten sind.
MPN-Schwerpunkt Ulm (Konstanze Döhner und Frank Stegelmann)
Ansprechpartner:
Prof. Dr. Konstanze Döhner und Prof. Dr. Frank Stegelmann
Universitätsklinikum Ulm
Klinik für Innere Medizin III
Albert-Einstein-Allee 23
89081 Ulm
Tel.: +49 731 500-45521
Fax: +49 731 500-45525
E-Mail: frank.stegelmann@uniklinik-ulm.de
Interessensgebiete:
(A) Molekulardiagnostik bei myeloproliferativen Neoplasien (MPN)Die molekulardiagnostische Aufarbeitung hat heutzutage bei MPN-Patienten einen wichtigen diagnostischen Stellenwert. Zunächst müssen BCR-ABL-negative MPN-Entitäten von der chronischen myeloischen Leukämie (CML) abgegrenzt werden. Die CML ist durch die Fusion der Gene BCR und ABL begründet, die sich aus der reziproken Translokation t(9; 22) ergibt. Auf molekularer Ebene kann eine BCR-ABL-Fusion mit Hilfe einer Multiplex-PCR auf cDNA-Ebene nachgewiesen bzw. ausgeschlossen werden. Nach Ausschluss einer BCR-ABL-Fusion sind bei V.a. eine myeloproliferative Neoplasie drei spezifische Genmutationen von Bedeutung, die Eingang in die Routinediagnostik gehalten haben und beim überwiegenden Teil der MPN-Patienten den Nachweis von Klonalität ermöglichen: Die Punktmutationen JAK2 V617F und MPL W515L sowie Frameshiftmutationen im Exon 9 des CALR-Gens. Es gibt unterschiedliche Nachweismethoden zur Identifizierung dieser drei Genmutationen im Labor (s. Abschnitt Labor). Häufig erfolgt der Nachweis aus Granulozyten-DNA. Die JAK2- und MPL-Mutation kann z.B. mittels einer allelspezifischen PCR unter Verwendung eines mutationsspezifischen Primers mit nachfolgender Genelektrophorese nachgewiesen werden. Für die Detektion einer CALR-Frameshiftmutation ist z.B. die Fragmentlängenanalyse aus Granulozyten-DNA eine gut geeignete Nachweismethode. Neben den drei genannten molekularen Markern ist bei speziellen Fragestellungen die Bestimmung zusätzlicher, seltenerer Genmutationen sinnvoll: Bei anhaltendem V.a. eine Polyzythämia vera ohne Nachweis von JAK2 V617F sollte mittels Sequenzieranalyse nach einer JAK2-Mutation im Exon 12 (z.B. der JAK2 K539L Mutation) gefahndet werden. Darüber hinaus sind das FIP1L1-PDGFRA-Fusionsgen und die KIT D816V-Mutation im Labor nachweisbare Mutationen bei V.a. chronische Eosinophilenleukämie bzw. Mastozytose.
(B) Quantifizierung der JAK2 V617F Mutation
Neben der molekulargenetischen Routineaufarbeitung in unserem Labor werden speziellere Analysen unter wissenschaftlichen Fragestellungen durchgeführt. Ein Interessensschwerpunkt ist dabei die quantitative Bestimmung der JAK2 V617F-Allellast bei Patienten mit nachgewiesener JAK2-Mutation im prospektiven Verlauf. Es ist nämlich bislang unklar, inwieweit die Allellast bei Patienten mit MPN Auswirkungen auf den klinischen Verlauf hat und mit einem erhöhten Risiko für vaskuläre Ereignisse oder eine fibrotische bzw. leukämische Transformation assoziiert ist. Für die Analysen wird aus Patientenblut (Granulozyten) DNA isoliert. Dann wird mittels quantitativer Real-Time PCR der Anteil mutierter Allele bestimmt. Diese Messungen werden im Erkrankungsverlauf wiederholt durchgeführt, um die Entwicklung der Allellast über die Zeit zu evaluieren und eventuelle Assoziationen mit dem klinischen Verlauf herzustellen. Darüber hinaus können auf diese Weise auch Bestimmungen minimaler Resterkrankung bei Patienten erfolgen, die sich einer intensiven Therapieform wie z.B. einer allogenen Stammzelltransplantation unterziehen. In Zukunft werden auch neuere Technologien wie z.B. die digitale PCR zur Quantifizierung der Allellast herangezogen.
(C) Detailierte genomische Charakterisierung von MPNNeben JAK2-, MPL- und CALR-Mutationen sind in den letzten Jahren weitere Mutationen bei myeloischen Neoplasien identifiziert worden, die auch bei einem Teil der Fälle mit MPN vorkommen und eine pathogenetische Bedeutung besitzen. Beispielhaft können hier Mutationen der Gene TET2, DNMT3A oderr ASXL1 genannt werden, die in der epigenetischen Regulation von Zielgenen eine wichtige Rolle spielen. Im Gegensatz zu Mutationen in JAK2/MPL/CALR sind diese weniger spezifisch für einzelne MPN-Entitäten, da sie auch bei MDS- und AML-Patienten vorkommen können. Außerdem besteht eine große Heterogenität der einzelnen Mutationen in disen Genen. Es kommen sowohl Punktmutationen als auch Frameshiftmutationen in unterschiedlichen Exons der Gene vor. Häufig fehlt eine „Hotspot“-Region. Diese Mutationen reflektieren so die genomische Komplexizität von MPN, sind wahrscheinlich für das heterogene klinische Bild der Erkrankungen verantwortlich und könnten auch bei der klonalen Entwicklung der Erkrankungen eine entscheidende Rolle spielen. Der klinische Stellenwert dieser Mutationen bei MPN ist nicht abschließend geklärt. In aktuellen Projekten waren daher homogene, klinisch gut charakterisierte MPN-Patientkohorten hinsichtlich des gleichzeitigen Auftretens von Mutationen, die über JAK2/MPL/CALR hinausgehen bzw. mit diesen zusammen vorkommen, untersucht. Dabei kommen neuere Techniken wie z.B. das ´Next-Generation-Sequencing´ zur Anwendung. Ziel ist es, eine Verbindung zwischen der molekularen Komposition der Patienten und den klinischen Charakteristika bzw. dem klinischen Verlauf und Therapieansprechen herzustellen. Die Daten sollen in Zukunft auch als Basis für funktionelle Analysen in Zellkultursystemen unter Einbeziehung der siRNA-Technik herangezogen werden.