Molekulargenetik Myeloproliferativer Neoplasien
Die molekulargenetische Charakterisierung myeloproliferativer Neoplasien (MPN) hat nicht nur zu einer erheblichen Verbesserung des pathogenetischen Verständnisses der Erkrankungen geführt, sondern auch Einzug in die diagnostische Aufarbeitung gehalten. Dadurch können die einzelnen Entitäten besser voneinander differenziert werden (Abb. 1).
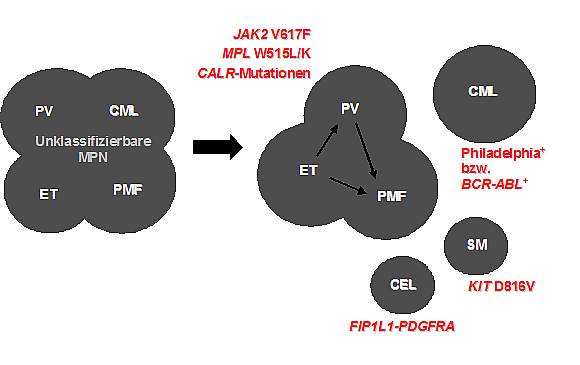
Abb. 1: Klassifikation der MPN im Wandel aufgrund der molekulargenetischen Charakterisierung (ET, essentielle Thrombozythämie; PV, Polyzythämia vera; PMF, primäre Myelofibrose; CML, chronische myeloische Leukämie; SM, systemische Mastozytose; CEL, chronische Eosinophilenleukämie).
BCR-ABL-positiv
Wichtig ist, die chronische myeloische Leukämie (CML) von den anderen MPN abzugrenzen. Die CML ist durch das Vorkommen des Philadelphila-Chromosoms, das durch eine Translokation zwischen den Chromosomen 9 und 22 zustande kommt [die sog. t(9;22)(q34;q11)], klar definiert und dadurch von den übrigen MPN abgrenzbar. Auf molekulargenetischer Ebene kommt es bei der CML zu einer Fusion der Tyrosinkinase ABL mit dem BCR-Gen, wodurch ein konstitutiv aktiviertes Bcr-Abl-Protein entsteht, das die unkontrollierte Myeloproliferation bei der CML begründet. Dies ist deswegen so bedeutsam, weil für die CML eine eigene Therapieform mittels sog. Tyrosinkinaseinhibitoren (z.B. Imatinib, Nilotinib oder Dasatinib) zur Verfügung steht, die die BCR-ABL-positiven Leukämiezellen zielgerichtet am Wachstum hemmen und zur Apoptose führen. Mit dieser Therapie können bei der CML anhaltend tiefe molekulare Remissionen erreicht werden, die die Prognose der Erkrankung sehr deutlich verbessert haben. Aufgrund der enormen klinischen Relevanz gehört der Nachweis bzw. Ausschluss einer BCR-ABL-Fusion bei den meisten MPN-Patienten zur diagnostischen Aufarbeitung mit dazu. Auf molekulargenetischer Ebene kann dafür eine sog. Multiplex-PCR mit anschließender Gelektrophorese herangezogen werden, mit die häufigsten BCR-ABL-Fusionstranskripte (e1a2, b2a2, b3a2) auf cDNA-Ebene nachgewiesen bzw. ausgeschlossen werden können (Abb.2).
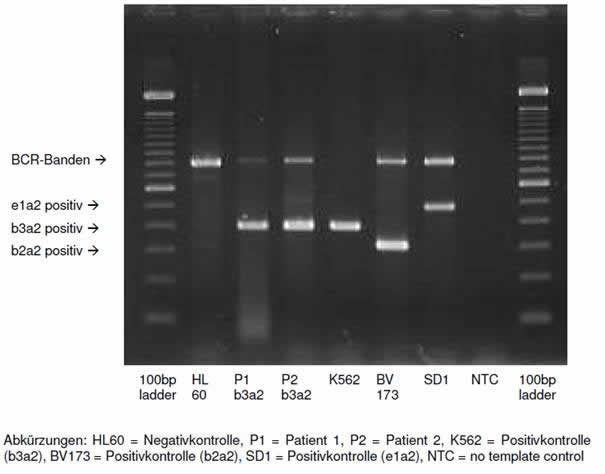
Abb. 2: Gelelektrophorese mittels Multiplex-PCR zum Nachweis bzw. Ausschluss von BCR-ABL-Positivität. Mit dieser Methode können die häufigsten BCR-ABL-Fusionstranskripte (e1a2, b2a2 und b3a2) erkannt werden.
BCR-ABL-negativ
Bei BCR-ABL-negativen MPN wie der essentiellen Thrombozythämie (ET), der Polyzythämia vera (PV) und der primären Myelofibrose (PMF) wurde in den letzten Jahren eine Vielzahl unterschiedlicher Genmutationen identifiziert. Von größter Bedeutung für die Routinediagnostik sind Punktmutationen in den Genen JAK2 (JAK2 V617F), MPL (MPL W515L/K) sowie Frameshiftmutationen im Exon 9 des CALR-Gens. Auf diese Weise kann bei annähernd allen PV-Patienten und bei 80-90% der PMF- bzw. ET-Patienten Klonalität nachgewiesen werden, wodurch die Diagnostik der MPN in den letzten Jahren erheblich erleichtert worden ist. Wichtig zu wissen ist dabei, dass die drei genannten Genmutationen in der überwiegenden Zahl der Fälle voneinander unabhängig vorkommen. PV-Patienten sind vor allem durch die JAK2 V617F Mutation (ca. 95%) und in selteneren Fällen (< 5%) durch Mutationen im Exon 12 des JAK2-Gens charakterisiert, während die V617F-Mutation bei etwa 50-60% der ET- und PMF-Patienten detektiert werden kann. In weiteren 20-30% der Fälle sind bei ET und PMF Frameshiftmutationen im CALR-Gen zu finden. Obwohl eine Vielzahl von unterschiedlichen Mutationen im Exon 9 des Gens lokalisiert, handelt es sich bei den meisten Patienten um eine sog. Typ-1 (52bp Deletion) oder Typ-2 Mutation (5bp Insertion), die zu einem neuen C-terminalen Peptidende des Proteins bei Patienten mit Mutation führen. MPL-Mutationen sind vergleichsweise am seltensten und kommen fast ausschließlich bei JAK2- und CALR-unmutierten ET- bzw. PMF-Patienten in einer Häufigkeit von ca. 5-10% vor. Hinsichtlich der funktionellen Auswirkungen geht man davon aus, dass nicht nur JAK2- und MPL-, sondern auch CALR-Mutationen zu einer JAK-STAT-Aktivierung mit der Folge einer unkontrollierten Zellproliferation führen.
Der molekulargenetische Nachweis von MPN-assoziierten Mutationen basiert auf der Polymerasekettenreaktion (PCR) von DNA (JAK2/MPL/CALR) bzw. cDNA (BCR-ABL). Ausgangsmaterial ist peripheres Blut. Im Falle der BCR-ABL-Diagnostik werden Leukozyten, für JAK2-/MPL-/CALR-Analysen in der Regel Granulozyten herangezogen. Es gibt eine Vielzahl unterschiedlicher Assays mit unterschiedlichen Sensitivitäts-Niveaus zum Mutationsnachweis. Eine verlässliche Methode für die JAK2-/MPL-Mutationsanalysen mit einer Sensitivität von ca. 0,1% stellte die sog. Allel-spezifische PCR dar. Dabei wird durch die gleichzeitige Verwendung von drei verschiedenen Primern eine Amplifikation der Wildtyp-Gensequenz als Kontrolle sichergestellt, während der dritte, mutationsspezifische Primer zu einer Amplifikation eines weiteren PCR-Produkts kürzerer Länge führt, falls eine Punktmutation nachweisbar ist. Zur Kontrolle müssen in der PCR eine bekannt-positive und eine bekannt-negative Probe mitgeführt werden. Hierfür bieten sich z.B. die Zelllinien HEL (JAK2 V617F mutiert) und HL-60 (JAK2 und MPL Wildtyp) an. Da für die MPL W515L-Mutation keine Zelllinie als Positivkontrolle zur Verfügung steht, kann hierfür ein bekannt positiver Patient mitgeführt werden, dessen Mutation zuvor mittels konventioneller PCR bestätigt worden ist (Abb. 3).
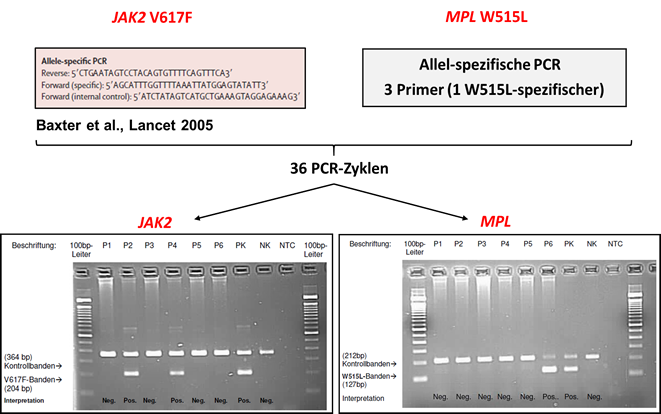
Abb. 3: Allel-spezifische PCR-Assays zum Nachweis der Mutationen JAK2 V617F (links) und MPL W515L (rechts) durch die Verwendung dreier Primer, von denen jeweils einer mutationsspezifisch ist.
Zum molekulargenetischen Nachweis einer CALR-Mutation werden in der Regel andere PCR-basierte Methoden als die Allel-spezifische PCR herangezogen. Eine mögliche Methode stellt zum Beispiel die Amplifikation des Bereichs von Exon 9 dar, in dem die Mutationen lokalisieren, mit anschließender sog. Fragmentlängenanalyse. Im Falle einer Mutation werden zwei unterschiedlich lange Fragmente amplifiziert, die aufgrund ihrer Größe von der Wildtypsequenz abgrenzbar sind. In der Fragmentlängenanalyse entsteht dadurch ein zweites Signal neben der Wildtyp-Kontrolle (Abb. 4). In einer darauffolgenden konventionellen Sanger-Sequenzierung kann die Mutation bestätigt und anhand gängiger Nomenklaturen genau benannt werden.
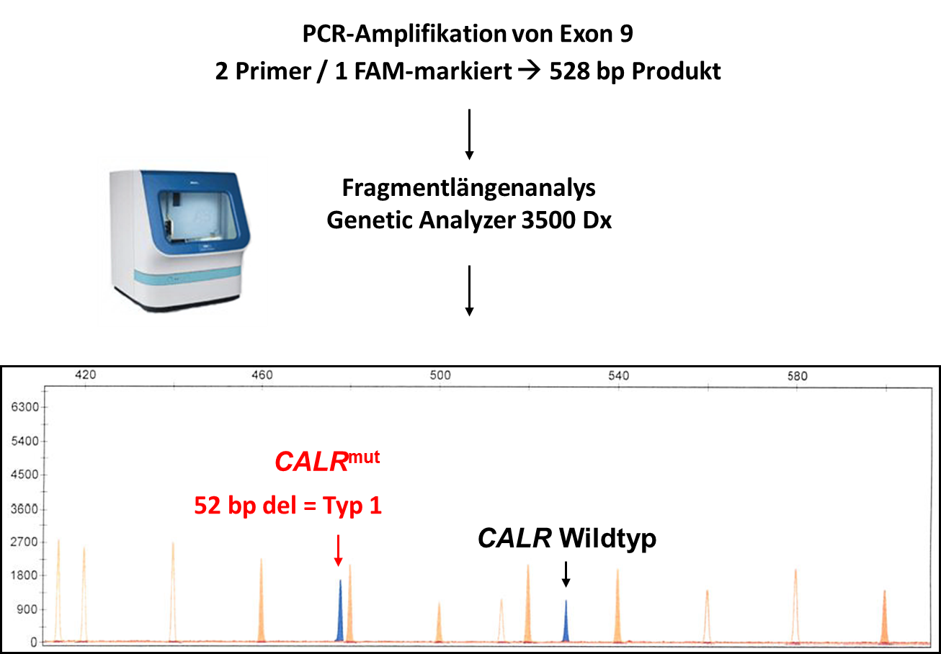
Abb. 4: Prinzip der CALR-Mutationsbestimmung am Beispiel der Fragmentlängenanalyse mit Nachweis einer Typ-1 Mutation (linker Peak).
Über die genannten drei genetischen Veränderungen hinaus sind in den letzten Jahren eine Reihe weiterer Mutationen bei BCR-ABL-negativen MPN-Patienten identifiziert worden. Die meisten davon betreffen epigenetische Regulationsgene wie z.B. TET2, ASXL1, EZH2 oder DNMT3A. Im Gegensatz zu JAK2-/MPL-/CALR-Mutationen sind diese Mutationen sehr heterogen. Es handelt sich dabei um eine Vielzahl unterschiedlicher Punkt- und Frameshiftmutationen. Innerhalb der Punktmuationen können neben Missense-Veränderungen wie bei der JAK2 V617F oder der MPL W515L/K Mutation auch sog. Nonsense-Mutationen vorkommen, die zu einem vorzeitigen Stopp der Translation und damit zu einem verkürzten Protein führen. Der biologische Stellenwert der meisten dieser Mutationen ist weniger gut definiert als bei JAK2, MPL und CALR. ASXL1-Mutationen können bei der Myelofibrose von ungünstiger prognostischer Bedeutung sein. Jedoch hat die Bestimmung von Mutationen, die über JAK2/MPL/CALR hinausgehen, noch keinen Eingang in die diagnostische Routine genommen und ist in den meisten Fällen vor allem speziellen, d.h. wissenschaftlichen Fragestellungen vorbehalten.